

従来、eCOAで患者からのデータ変更を反映させることは難しい場合がありました。その主な理由は、変更の確認や承認プロセスが明確でなかったこと、また使用されているシステムにデータ編集機能が備わっていなかったことにあります。
この問題は、患者、施設スタッフ、CRA、サービスプロバイダー、スポンサーなど、すべての関係者に課題をもたらしました。各利害関係者は、それぞれのプロトコル、GCP(治験実施基準)、規制の範囲内で義務を果たすよう努めていますが、役割と責任が十分に明確化されていないことが、さらなる困難を引き起こしています。
この記事では、電子患者報告アウトカム(ePRO)データの変更に関する指揮命令系統の確立からデータ収集に至るまで、その背景を考察し、包括的な新たなベストプラクティスを提言した最近発表された論文を紹介します。

臨床試験データは、全ての参加者間で回答オプションが標準化されるよう、検証済みの質問票を使用して収集されます。疾患症状や治療の副作用から、身体的、社会的、感情的、認知的な機能的アウトカムに至るまで、患者が報告するデータは意思決定に役立ち、最終的には医療政策や実践に影響を与えます。
eCOAは、従来の手作業による紙ベースの評価に比べ、より正確かつ効率的なデータ収集方法です。たとえば、Viedoc MeのようなePRO/eCOAモジュールを使用すると、患者のスマートデバイスから直接データを取得でき、データの質の向上と患者コンプライアンスの強化が可能になります。

現在の規制では、施設とスポンサーの責任が明確に分けられています。施設は原資料の管理とその維持を担当し、スポンサーはプロトコールに従って治験の実施を監督する役割を持ちます。
規制要件では、原資料の完全性と正確性を維持するために、データの変更や修正には必ず日付、イニシャル(署名)、および理由を記載する必要があるとされています。
ミスが発生することは避けられませんが、特にePROデータの変更が記憶バイアスに起因する可能性がある場合、その変更は慎重に管理される必要があります。たとえば、患者がある症状や副作用が「最初に思ったほど悪くなかった」と考え直し、記録した翌日に回答を変更するケースなどです。これは懸念事項といえます。なぜなら、痛みの記憶は正確でないことが多いため、このような変更に対しては慎重に対応する必要があります
一方で、例えば施設のコーディネーターが誤って別の患者のデータとして入力し、すぐにそのミスに気づいた場合、そのデータを迅速に正しい場所へ移す必要があります。
業界では、DCR(データ変更要求)を提出、評価、議論、実施するためのスケーラブルなプロセスが長い間求められてきました。しかし、明確な指針が不足しているため、多くのサービスプロバイダーはスポンサーと協議の上、患者が入力したデータの変更を一切認めない方針を取っていました。しかし、規制当局はこのアプローチは受け入れられないと示唆しています。

Viedocの薬事マネージャーであるAlan Yeomansは、最近、PROデータの変更実行およびドキュメント化のプロセスを詳細に検討するために集まった、 Critical Path Institute のPROコンソーシアム、eCOAコンソーシアム、eClinicalフォーラムの専門家チームに参加しました。
その結果、2023年12月にSCDMのウェブサイトで公開された論文「臨床結果評価データの変更に関するベストプラクティス推奨事項1」が発表されました。この論文では、合意された役割と責任に基づいた試験固有のプロセスについて、長く待たれていた概要が示されています。また、治験責任医師がデータの正確性を維持し、変更が明確な根拠と文脈によって裏付けられるよう設計されたワークフローについても説明されています。
「私たちは、データ変更に関する優れたプロセスを構築しようと考えましたが、それが容易ではないことにすぐ気づきました。何度も議論を重ねる中で、基本原則を定義する必要性を感じました。そして、PROデータの変更の取り扱い方法を明確に規定する監督計画、すなわちデータ管理計画が必要であるとの結論に至りました。」と Alanは次のように振り返ります。
Alanはさらに次のように説明します。「施設は常にデータを管理下に置く必要があります。データに責任を持つのは施設です。一方で、スポンサーは試験の監視を行い、何が起こっているのかを把握しなければなりません。
データ変更によってALCOA+の遵守が損なわれることがあってはなりません。そのため、データ変更時にも、当初のデータ収集時と同じ規制をすべて遵守する必要があります。また、すべての変更は監査証跡によって完全に記録されなければなりません。」

監督計画を策定する際には、あらゆる変更依頼シナリオに対応する明確なワークフローを確立する必要があります。この計画では、各変更タイプに関連する手続きおよび技術的な詳細を定義する必要があります。また、どのDCRタイプが重要であるか(施設へのフォローアップ、スポンサー/CROへの通知や協議、またはその両方を必要とする変更)と、どのタイプが手続き上のものか(フォローアップを必要としない)も明確に定義します。
まず、プロトコルデータをクリティカルデータポイントとプロシージャルデータポイントに分類し、それらを監視計画に記載することが重要です。計画では、プロトコルとの整合性を確保し、データの完全性を維持することに加え、原資料に対する施設の管理責任や治験依頼者による治験監視に関する規制要件を満たす形で、各データ変更プロセスを概説する必要があります。
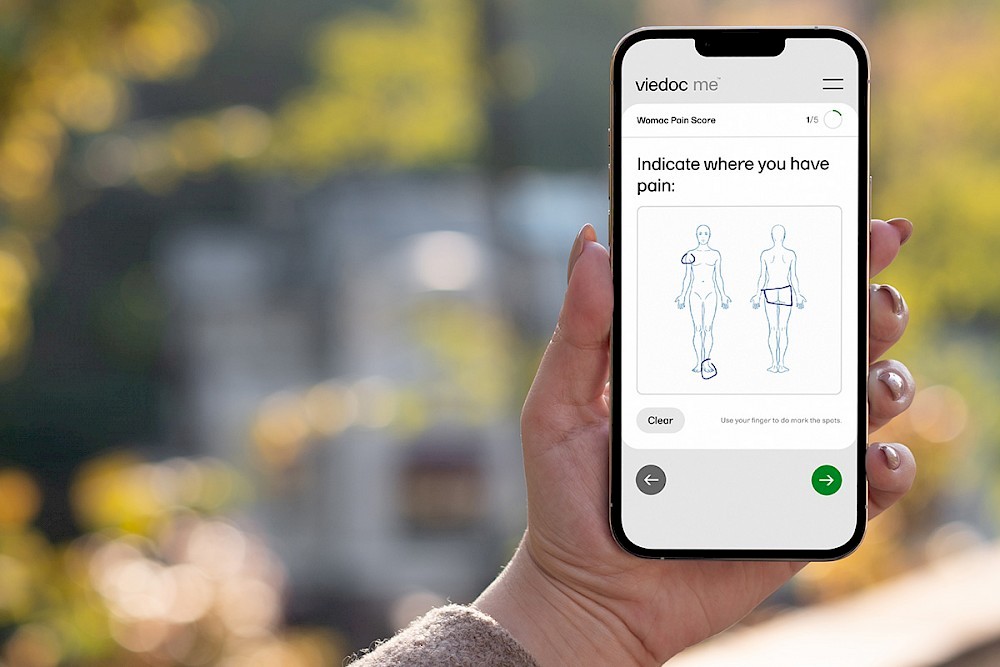
Viedoc Meは、Viedocシステムと統合されたePRO/eCOAモジュールで、患者がスマートフォン、タブレット、またはコンピュータを使用してプラットフォームに直接データを入力できるように設計されています。これにより、データ収集の効率と正確性が向上し、患者の来院回数を減らすことが可能です。
また、治験が円滑に進行することで、患者にとってより良い体験となり、治験への継続参加の可能性が高まります。さらに、自身の端末でデータを入力できることで、患者はコントロール感を得られ、治験への積極的な参加が促されます。
Viedoc MeをeCOAソリューションとして使用する利点
より迅速なデータ収集
患者から直接データを収集することで、データ入力や転記エラーを排除します。
統合ソリューション
患者に送信するフォームや、CRFの一部となるフォームをユーザーが設定可能です。
ファイルのアップロード
患者はスマートフォンから画像ドキュメントを直接送信できます。
患者の積極的な参加
ユーザーフレンドリーなインターフェースにより、患者の積極的な参加とコンプライアンスの向上が促進されます。
リアルタイムのデータ収集
リアルタイムのデータ収集が容易になり、臨床試験データの質と適時性が向上します。
Viedoc、特にViedoc Meが貴社の臨床試験にどのような変革をもたらすかについて、詳しく知りたい方は、ぜひデモを予約してください。